
Hemostasis
last authored: Nov 2009
last reviewed:
Introduction
Hemostasis is a set of well-regulated processes designed to rapidly and specifically form a clot at sites of vascular injury, such as occurs in a cut. Elaborate mechanisms are also in place to limit the clot and maintain blood in a fluid, clot-free state in normal vessels. The pathologic manifestation of hemostasis is thrombosis - a blot clot forming in uninjured or mildly injured vessel.
After injury, there is an immediate and brief period of arteriolar vasoconstriction mediated by reflex neurogenic mechanisms and augmented by secretion of factors such as endothelin.
Wound healing follows hemostasis.
Mechanisms of Hemostasis
Hemostasis involves two different mechanisms - platelet aggregation and coagulation.
- platelet plug
- coagulation cascade
Platelet Plug
In the seconds following endothelial injury, the highly thrombogenic extracellular matrix binds platelets and leads to their activation. von Willebrand factor (vWF) is an essential cofactor for platelet binding. Platelets undergo a shape change and release secretory granules. This recuits other platelets, and within minutes a hemostatic plug forms. Erythrocytes and leukocytes also are found in hemostatic plugs.
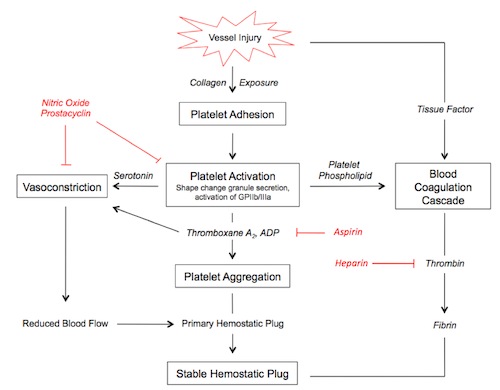
illustration by Kathryn Dorman
When circulating, platelets are smooth discs with a number of integrins - membrane bound glycoprotein receptors - on their surface. They also contain two types of granules full of clot-forming proteins.
Alpha granules have P-selectins on their membranes and contain fibrinogen, fibronectin, factors V and VIII, platelet factor 4, platelet-derived growth factor, and TGFβ.
Dense, or δ granules, containe adenine nucleotides ADP and ATP, ionized calcium, histamine, seratonin, and epinephrine.
Platelet contact with ECM components such as collagen, proteoglycans, fibronectin, and other adhesion glycoproteins leads to three general reactions: adhesion and shape change, secretion, and aggregation.
Platelet adhesion is largely mediated by interactions with vWF, acting as a bridge, together with clotting factors, between membrane-bound glycoprotein Ib and collagen.
Secretion of both granule types occurs soon after adhesion, mediated by a phosphorylation cascade. ADP is a potent mediator of platelet aggregation. Phospholipid complexes are also expressed at the cell surface, providing critical nucleation sites for the intrinsic clotting cascade.
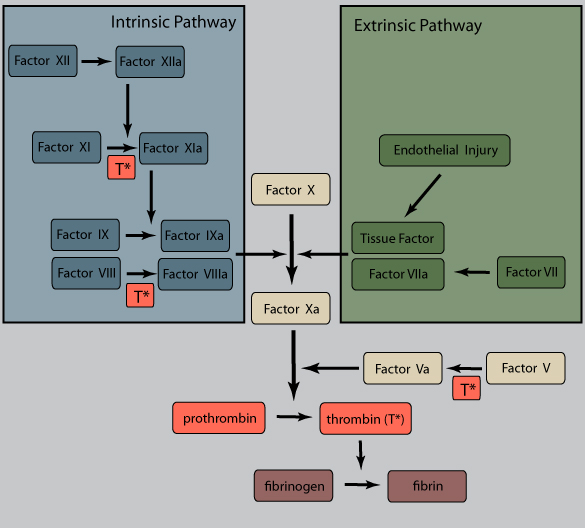
Platelet aggregation is mediated by platelet release of ADP and thromboxane A2 (TxA2). This sets up an autocatalytic reaction whereby the aggregation enlarges to create a hemostatic plug. Activation of the coagulation cascade liberates thrombin, which converts fibrinogen into fibrin in and around the platelet plug, cementing things in place.
Noncleaved fibrinogen can also cause platelet aggregation via cell surface GpIIb-IIIa.
Endothelial prostacyclin (PGI2) and nitric oxide (NO) inhibit platelet aggregation. PGI2 functions by increasing levels of platelet cAMP, inhibiting activation and aggregation. PGI2 and NO act as vasodilators.
Antihemostasis
Hemostasis is carefully limited to site of injury by various mechanisms.
Antiplatelet Effects
An intact endothelium prevents platelet contact with the thrombogenic ECM. Endothelial prostacyclin (PGI2) and nitric oxide (NO), both of which are potent vasodilators, prevent platelet binding to uninjured endothelium. Their synthesis is mediated by a number of factors, including thrombin and various cytokines produced during coagulation. Endothelial cells also produce adenosine diphosphatase, degrading ADP and inhibiting platelet aggregation.
Anticoagulant Effects
Membrane associated heparin-like molecules interact with antithrombin III to inactivate thrombin, factor Xa, and several other factors. Antithrombin III's effectiveness is increased 1000x through binding to heparan sulfate, a sugar found in the endothelial lumen.
Thrombomodulin binds to thrombin, preventing it from producing fibrin and converting it to an anticoagulant molecule by activating protein C. Protein C, which also requires protein S, inhibits clotting by degrading factors Va and VIIIa. Inflammation, pregnancy, contraceptives, liver disease, or HIV can decrease protein C and S levels.
Tissue factor pathway inhibitor (TFPI) is a plasma serine protease inhibitor which complexes and inhibits activated factor VIIa and Xa.
Fibrinolytic Effects
Endothelial cells synthesize tissue- plasminogen activator (t-PA), a potent enzyme which cleaves plasminogen to form active plasmin, which clears fibrin deposits from endothelial surfaces. tPA's activity is greatly increased by binding to fibrin.
Prostacyclin is synthesized and secreted by endothelial cells, increasing platelet levels of cAMP.
Clot Resolution
Hemostasis and Disease
In people with protein C or S deficiency, warfarin can induce a hypercoagulable state
Resources and References
 |